Importancia de los estudios de estabilidad de medicamentos. Consideraciones generales
Q.F. Angelina Salinas Reyes
Laboratorios Saval S.A.
El objetivo de este artículo es describir las diferentes aplicaciones de los estudios de estabilidad en la industria farmacéutica y describir los tipos de estudios que los fabricantes necesitan llevar a cabo para poder comercializar sus productos en diferentes mercados.
El propósito de los estudios de estabilidad es establecer cómo la calidad de un principio activo o producto farmacéutico puede variar en el tiempo, bajo la influencia de factores ambientales como la temperatura, la humedad y la luz. Estos estudios permiten así, definir las condiciones de almacenamiento, los materiales de envase y/o embalaje, y los períodos de reanálisis y vida útil.
La estabilidad depende tanto de los factores ambientales mencionados como de factores propios del producto: propiedades físicas y químicas del principio activo y de los excipientes farmacéuticos, forma farmacéutica, proceso de fabricación, naturaleza del sistema de cierre del envase, además de las propiedades de los materiales del envase.
Estudios a corto plazo o acelerados:
Son estudios de tipo predictivo y por tanto provisionales, aplicados al desarrollo de nuevos productos.
Se desarrollan aplicando condiciones que aumentan la velocidad de degradación o los cambios físicos de un principio activo o de un producto farmacéutico a partir de pruebas de almacenamiento severas en cuanto a temperatura y humedad.
Permiten proponer la vida útil de un producto o evaluar el efecto de un cambio crítico.
Estudios de largo plazo o prolongados:
Son estudios de tipo confirmatorio y por tanto definitivos, realizados por el tiempo total del período de eficacia propuesto. Se realizan en condiciones ambientales controladas y correspondientes con las condiciones climáticas de la zona donde el producto será comercializado.
Los estudios de estabilidad se realizan en distintas etapas del ciclo de vida de un producto farmacéutico.
En la fase de pre-formulación y desarrollo de producto:
Las pruebas aceleradas de estabilidad constituyen un medio para comparar diferentes formulaciones, materiales de envase o procesos de fabricación en experimentos de corta duración. Tan pronto como se establecen la formulación y el proceso de fabricación definitivos, el fabricante lleva a cabo una serie de pruebas de estabilidad aceleradas que permiten predecir la estabilidad del producto y determinar su tiempo de vida útil junto con las condiciones de almacenamiento. Paralelamente se inician los estudios en tiempo real o prolongados para fines de confirmación.
Estudios de estabilidad para registro sanitario y autorización de comercialización:
Es requisito regulatorio presentar la información generada mediante estudios de estabilidad acelerados y a tiempo real o prolongados, en la forma farmacéutica definitiva y en el material de envase y empaque definitivo.
Estudios de estabilidad durante la comercialización:
El fabricante está obligado a realizar estudios de estabilidad en tiempo real de acuerdo con la normativa vigente en el país donde se aprueba el registro sanitario para confirmar la fecha de vida útil y las condiciones de almacenamiento previstas con anterioridad.
Además, una vez que un producto ha sido registrado, cada vez que modifica o amplia el origen del fabricante de la sustancia activa, o se hacen modificaciones importantes de la formulación, o cambia el proceso de fabricación, especificaciones y metodologías analíticas, entre otros, se requiere realizar los respectivos estudios de estabilidad y los resultados deben ser comunicados a la autoridad sanitaria.
Estudios de estabilidad “on going” post- comercialización:
Todos los productos en el mercado deben ser monitoreados a través de un programa continuo para demostrar que se mantiene la estabilidad y calidad durante toda su vida útil. Esto aplica a cada producto en sus diferentes dosis y tipo de envase primario y embalaje; es un programa continuo que establece retener usualmente un batch por año y someter dichos productos a estudios de largo plazo en las condiciones de almacenaje establecidas en sus rótulos.

Guías para la realización de estudios de estabilidad
Dentro de la industria biofarmacéutica, la determinación de la estabilidad de productos y principios activos es parte integral de su proceso de desarrollo y debe realizarse de acuerdo con exigencias regulatorios específicas. Deben emplearse estrictos métodos de prueba para determinar estabilidad, seguridad, integridad y vida útil de los productos en una variedad de condiciones.
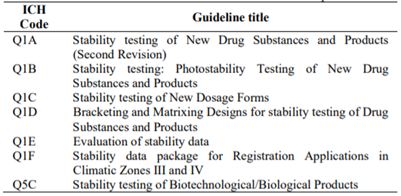
Para comercializar, los fabricantes deben realizar y presentar los resultados de estabilidad de los principios activos y del producto terminado al momento de solicitar el registro sanitario o la autorización de comercialización; por ello existen organismos internacionales especializados que elaboran guías o lineamientos que luego las autoridades sanitarias de cada país adoptan como normas exigibles. Así los fabricantes pueden asegurar que los pacientes recibirán medicamentos de calidad, eficaces y seguros. (Tabla 1)
 El ICH es el Consejo Internacional para la Armonización de requisitos técnicos para el registro de medicamentos de uso humano. Se trata de un proyecto que reúne a las autoridades reguladoras de Europa, Japón y Estados Unidos, y en él, expertos farmacéuticos de estas tres regiones discuten y consensuan aspectos científicos y técnicos para el registro de productos. Su objetivo es mejorar, por medio de la armonización de sus reglamentos, el proceso de desarrollo y registro de nuevos medicamentos en Europa, Japón y Estados Unidos. Las directrices de la ICH se han adoptado como ley en muchos países. En Chile, la norma técnica N°129 ‘‘Guía para la Realización y Presentación de Estudios de Estabilidad de Productos Farmacéuticos” establece los lineamientos para la ejecución de estudios de estabilidad tomando como referencia la directriz ICH / OMS. (www.ispch.cl).
El ICH es el Consejo Internacional para la Armonización de requisitos técnicos para el registro de medicamentos de uso humano. Se trata de un proyecto que reúne a las autoridades reguladoras de Europa, Japón y Estados Unidos, y en él, expertos farmacéuticos de estas tres regiones discuten y consensuan aspectos científicos y técnicos para el registro de productos. Su objetivo es mejorar, por medio de la armonización de sus reglamentos, el proceso de desarrollo y registro de nuevos medicamentos en Europa, Japón y Estados Unidos. Las directrices de la ICH se han adoptado como ley en muchos países. En Chile, la norma técnica N°129 ‘‘Guía para la Realización y Presentación de Estudios de Estabilidad de Productos Farmacéuticos” establece los lineamientos para la ejecución de estudios de estabilidad tomando como referencia la directriz ICH / OMS. (www.ispch.cl).
Tabla 1: Códigos y títulos usados en la Guías ICH para productos humanos
El diseño de los estudios de estabilidad debe considerar la zona climática del mercado previsto para su comercialización. Así, en todo el mundo se distinguen 4 zonas climáticas que difieren en cuanto a condiciones de temperatura y humedad, resumidas en la Tabla 2. Las condiciones de humedad relativa (%HR) y temperatura (ºC), pueden ser simuladas en cámaras o salas de estabilidad (Fig. 1), es decir en sistemas cerrados que permiten mantener estables dichos parámetros.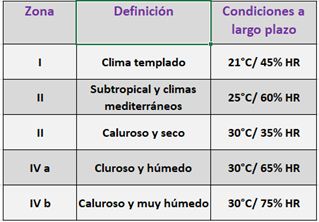
Fig. 1 Cámaras / Salas Climáticas para realizar Estudios de Estabilidad

A continuación, se describe el caso general de las condiciones de almacenamiento con propósitos de solicitud de registro de producto. Tabla 3
Adicionalmente, es deseable estudiar los efectos del transporte y los ciclos de temperatura, para demostrar que no habrá eventos imprevistos durante el envío y el almacenamiento. Tabla 3

Las pruebas de estabilidad en uso se aplican a productos de dosis múltiples. El objetivo es simular el uso del producto en la práctica normal, siguiendo las instrucciones de reconstitución, y también establecer un período de tiempo durante el cual se puede usar un producto una vez que el envase se ha abierto.
Rol del material envase y empaque para la estabilidad del producto farmacéutico:
El empaque es un componente muy importante y se selecciona durante el desarrollo de los medicamentos; de no ser adecuado, puede afectar la estabilidad y seguridad de los medicamentos. El material de envase se elige según su eficacia para preservar la calidad, potencia y seguridad de los productos farmacéuticos.

La FDA (Food and Drug Administration) de los Estados Unidos define el sistema de envase y cierre (container closure system) como la suma de los materiales de acondicionamiento que contienen y protegen la forma galénica.
El cierre de un envase farmacéutico forma parte del acondicionamiento primario y, como tal, contribuye a garantizar la estabilidad y la inviolabilidad del contenido. Al formar parte del acondicionamiento primario y estar en contacto con el producto, el cierre debe sellar el envase de forma segura, pero sin reaccionar con él producto (de lo contrario podría alterar su composición química y/o su estructura física).
Control de la Humedad para alcanzar la estabilidad
La humedad es uno de los mayores desafíos que un fabricante de medicamentos enfrenta. Algunos principios activos (PA o API) son altamente sensibles a la humedad. Este hecho, junto con otros factores ambientales, da cuenta de un riesgo importante para la estabilidad de los productos. La información de materiales de envase y sus propiedades (como la tasa de transmisión de vapor de agua: WVTR), constituye un factor clave a considerar para garantizar la estabilidad de un producto durante toda su vida útil.
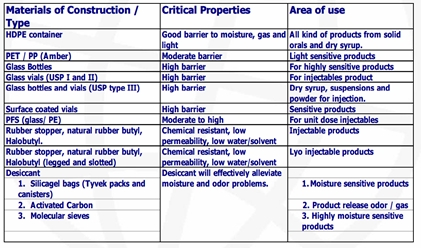
Para ilustrar la diversidad de materiales y su naturaleza, se adjuntan las Tablas 4 y 5, referidas en J Bhat IPA seminar ppt Packaging role in stability .ppt (Jnanadeva Bhat M.)
Tabla 4. Tasa de transmisión de vapor de agua (WVTR) de varios materiales blíster film.
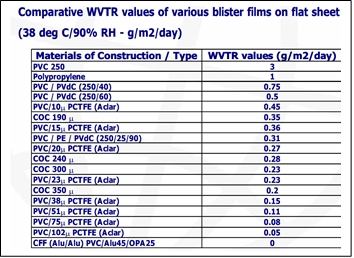
Tabla 5. Propiedades de envases y cierres
Importancia de dar cumplimiento a las condiciones de almacenamiento recomendadas
Las condiciones ambientales recomendadas se incluyen en las etiquetas y/o estuches de los productos farmacéuticos y es imprescindible respetarlas. Pueden especificar un intervalo de temperatura específico o un lugar determinado de almacenamiento, por ejemplo, “en el refrigerador” o “almacenado a no más de 30°C”, u otras instrucciones complementarias como “proteger de la luz”. La naturaleza del material de envase primario (en contacto con el producto) se define mediante estudios de estabilidad. Se recomienda no quitar el material de protección hasta haber utilizado el contenido en su totalidad.
Algunos medicamentos, la mayoría de las vacunas, insulinas y algunos productos oftálmicos entre otros, deben conservarse a temperaturas de refrigeración, es decir, deben mantener una “cadena de frío” para asegurar su integridad, calidad y seguridad. La autoridad sanitaria en Chile (www.ispch.cl) establece a través de su Norma Técnica N° 208 (09/2019), los requisitos específicos para el almacenamiento y transporte de medicamentos refrigerados y congelados, aplicables a cámaras de frío, refrigeradores, congeladores, vehículos móviles de transporte, contenedores fríos y termos magistrales.
La condición de “cadena de frío” consiste en una línea de suministro con temperatura controlada, es decir una serie ininterrumpida de actividades y condiciones para la fabricación, almacenamiento, transporte, distribución y venta de productos en un rango entre 2° y 8°C, dado que un “no cumplimiento” en cualquier nivel de esta cadena implica un riesgo de pérdida de atributos de calidad y, por ende, un potencial impacto sobre la salud de los pacientes.
En conclusión, sobre el fabricante de productos farmacéuticos recae la gran responsabilidad de establecer condiciones de almacenamiento para garantizar la vida útil recomendada para cada producto y en cada país de destino, siempre con base en estudios de estabilidad. Por esta vía el fabricante garantiza el mantenimiento de la calidad, inocuidad y eficacia a lo largo del tiempo de almacenamiento.
En la mantención de las condiciones de almacenamiento, intervienen distintos responsables según la etapa del ciclo de vida del producto: fabricante, distribuidores y el usuario final. Este último necesita estar informado de cómo hacer un buen uso y manipulación del producto.
Bibliografía:
1.-International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines (http://www.ich.org)
ICH Q1A (R2): Stability testing of new drug substances and products.
ICH Q1B: Photostability testing of new drug substances and products.
ICH Q1C: Stability testing of new dosage forms.
ICH Q1D: Bracketing and matrixing designs for stability testing of new drug substances and products.
ICH Q1E: Evaluation for stability data.
ICH Q5C: Quality of biotechnological products: stability testing of biotechnological/biological products.
2.-Stability testing of active pharmaceutical ingredients and finished pharmaceutical products. In: WHO Expert Committee on Specifications for Pharmaceutical Preparations: forty-third report. Geneva: World Health Organization; 2009: Annex 2 (WHO Technical Report Series, No. 953).
3.-Stability testing of (api) active pharmaceutical ingredients and finished pharmaceutical products: WHO TRS (Technical Report Series) 1010, 2018 Annex 10.
4.-Norma Técnica N° 129 / 2012 “Guía para la realización y presentación de los estudios de estabilidad de los productos farmacéuticos en Chile y su Anexo”
5.-Stability Studies and Testing of Pharmaceuticals: An Overview JUNE 2020 LCGC NORTH AMERICA VOLUME 38 NUMBER 6 ( www.chromatographyonline.com)
6.-Key Considerations in Stability Testing; Felicity Thomas; Pharmaceutical technology march 2019 PharmTech.com
7.-Stability Testing: The Crucial Development Step; Felicity Thomas; Pharmaceutical Technology MARCH 2020 PharmTech.com
8.-Ongoing Stability Testing: Requirements, Solutions and Potential Pitfalls; Innovations in Pharmaceutical Technology; By Sven Oliver Kruse at Diapharm Analytics GmbH
9.-Role of packaging material on Pharmaceutical product stability; Jnanadeva Bhat M.; DGM - Product Developmen; Associated Capsules Pvt Ltd
10.- Norma Técnica N° 208 “Almacenamiento y transporte de medicamentos refrigerados y congelados”, 17.09.2019.